MIPdatabase
MIPdatabase



Authors: Wang SS, Yang BW, Zhu QJ
Article Title: Configurational Simulations and Theoretical Calculations of Molecularly Imprinted Polymers of Histamine and 2-(Trifluoromethyl)acrylic Acid Based on Computational Chemistry.
Publication date: 2017
Journal: Journal of the Chinese Chemical Society
Volume: 64
Issue: (4)
Page numbers: 434-439.
DOI: 10.1002/jccs.201600282
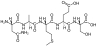
Abstract: In this article, we provide a theoretical discussion on the interactions between a template molecule and functional monomer(s) in the preparation of molecularly imprinted polymers (MIPs). Density functional theory (DFT) was used to compute the 3D structures, natural bond orbital, and binding energy in the template-monomer(s) complexes. Histamine (HA) and 2-(trifluoromethyl)acrylic acid (TFMAA) were, respectively, selected as the template and the monomer. The computational process was performed according to B3LYP method with 6-311 + (d,p) basis set under the different HAGÇôTFMAA ratios from 1:1 to 1:10. The computational results show that the HA-TFMAA complex at the ratio of 1:5 yields no consequence. Furthermore, the HA-TFMAA complex at the ratio of 1:5 allowed the minimum binding energy and the steadiest condition, with four hydrogen bonds. Configurational simulations and theoretical calculations of the template-monomer complex can be used as an appropriate guiding tool for manufacturing MIPs with high specificity and selectivity, thereby avoiding repeated experiments and wastage of substantial time and investment
Template and target information: histamine, HA
Author keywords: histamine, 2-(trifluoromethyl)acrylic acid, Molecularly imprinted polymers, Computational chemistry


Join the Society for Molecular Imprinting
New items RSS feed
Sign-up for e-mail updates:
Choose between receiving an occasional newsletter or more frequent e-mail alerts.
Click here to go to the sign-up page.
Is your name elemental or peptidic? Enter your name and find out by clicking either of the buttons below!