MIPdatabase
MIPdatabase



Authors: Zhang TQ, Zhang W, Liu L, Chen Y
Article Title: Simultaneous detection of site-specific histone methylations and acetylation assisted by single template oriented molecularly imprinted polymers.
Publication date: 2020
Journal: Analyst
Volume: 145
Issue: (4)
Page numbers: 1376-1383.
DOI: 10.1039/C9AN02360G
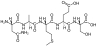
Abstract: Histone post-translational modification (PTM) is a marker for gene transcription and is involved in a range of cancers, such as breast cancer. Most importantly, different modifications of a specific site (e.g., mono-, di- and tri-methylations and acetylation at a lysine residue) are individually enriched in particular regions of the genome, indicating that they may serve distinct functions. How these patterns are built and whether downstream events are dependent on the precise site-specific modification status are still poorly understood. One of the prerequisites is to obtain quantitative information on individual modification forms. Currently, the low abundance of these forms creates a great challenge. In this study, a targeted proteomics assay based on liquid chromatography-tandem mass spectrometry (LC-MS/MS) coupled with single template oriented molecularly imprinted polymers (MIPs) was developed to attempt to simultaneously quantify site-specific histone mono-, di- and tri-methylated and acetylated forms. Surrogate peptides, including an unmodified one, were selected as targets in both MIP enrichment and subsequent LC-MS/MS detection. As a consequence, a limit of quantification (LOQ) of ~0.5 nM was achieved, which was 10-fold more sensitive than without MIPs and much more sensitive than mass spectrometry involving a scanning mode. Finally, the assay was employed to quantitatively analyze the H3K79, H3K122 and H4K31 modification status in human breast cell lines
Template and target information: peptide, methylated histone, acetylated histone, histone, H3K79, H3K122, H4K31


Join the Society for Molecular Imprinting
New items RSS feed
Sign-up for e-mail updates:
Choose between receiving an occasional newsletter or more frequent e-mail alerts.
Click here to go to the sign-up page.
Is your name elemental or peptidic? Enter your name and find out by clicking either of the buttons below!