MIPdatabase
MIPdatabase



Authors: Pavel D, Lagowski J
Article Title: Computationally designed monomers and polymers for molecular imprinting of theophylline - part II.
Publication date: 2005
Journal: Polymer
Volume: 46
Issue: (18)
Page numbers: 7543-7556.
DOI: 10.1016/j.polymer.2005.05.146
Alternative URL: http://www.sciencedirect.com/science/article/B6TXW-4GFNG6N-B/2/3b51050c4ee52d8fd9b0c7492317ca26
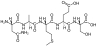
Abstract: The main objective of this research is to develop and apply state-of-the-art computational tools to achieve an understanding of intermolecular interactions in molecular imprinting of theophylline into complex polymeric systems. Molecular dynamics (MD) simulations were carried out for different molecular systems in order to predict the interaction energies, the closest approach distances and the active site groups between the simulated molecular systems and different bio-ligands. The minimized structures of five ligands, theophylline and its derivatives (theobromine, theophylline-8-butanoic acid, caffeine and theophylline-7-acetic acid) have been obtained with the use of molecular mechanics approach. NVT MD simulations at room temperature were carried out to obtain equilibrated conformations in all cases.The first simulated molecular systems consisted of a ligand and commonly used functional monomers, polymers and a substrate. The second simulated molecular systems consisted of a ligand and a monomer or polymer using a solvent (ethanol).During this study, it was found that electrostatic interactions play the most significant role in the formation of molecular imprinting materials. The simulated functional monomers and polymers with ligands indicate that the functional groups interacting with ligands tends to be either -COOH or CH2CH-. It was also found that molecular substrate without functional side groups are recommended for molecular imprinting technology. For both the solvated monomer and polymer systems is that it appears that the presence of the solvent appears to favour the formation of more stable clusters with theophylline than with its derivatives
Template and target information: theophylline
Author keywords: imprinting polymers, ligand, Molecular simulation



Join the Society for Molecular Imprinting
New items RSS feed
Sign-up for e-mail updates:
Choose between receiving an occasional newsletter or more frequent e-mail alerts.
Click here to go to the sign-up page.
Is your name elemental or peptidic? Enter your name and find out by clicking either of the buttons below!